


Case Studies
Case Studies
- Preparation of Hyperbranched Resin and Its Effect on the Performance of Epoxy Resin Channeling Control Agent (Part 1)
- Preparation of Hyperbranched Resin and Its Effect on the Performance of Epoxy Resin Channeling Control Agent (Part 2)
- Construction of A New Organic-inorganic Composite Emulsion and Its Enhanced Mechanical Properties of Oil Well Cement(Part 1)
- Construction of A New Organic-inorganic Composite Emulsion and Its Enhanced Mechanical Properties of Oil Well Cement(Part 2)
- The Application Prospects of DeepSeek Large Model in Petroleum Engineering(Part 1)
- The Application Prospects of DeepSeek Large Model in Petroleum Engineering(Part 2)
- Development and Performance Evaluation of Efficient Asphalt Dispersant (Part 1)
- Development and Performance Evaluation of Efficient Asphalt Dispersant (Part 2)
- Development and Performance Evaluation of Efficient Asphalt Dispersant (Part 3)
- Research and Application Status of Drilling Fluid Plugging Materials (Part 1)
.png)
Polyacrylamide has a wide range of applications in petroleum production, and it is of great significance to study the properties of polyacrylamide solution and the mechanism of its application at the microscopic level. In recent years, computer molecular simulation technology has been continuously developing, which has high flexibility and fills the gap between macroscopic experiments and theory. It is increasingly being applied to the study of molecular behavior in polymer systems. This article reviews the research progress of computer molecular simulation technology in the application of polyacrylamide in oil fields, elaborates on the main molecular simulation technologies and application types, and reveals the beneficial role of this technology in the field of petrochemicals.
Polyacrylamide (PAM) is a linear water-soluble polymer formed by homopolymerization of acrylamide or copolymerization with other monomers. PAM is currently widely used in fields such as oil extraction, sewage treatment, food processing, and medicine, and is known as a "versatile additive". In recent years, China's PAM production has reached one million tons, of which the demand for increased oil production accounts for 44.9%, making it the largest downstream market. Especially in the fields of profile control and water plugging, tertiary oil recovery, and drilling, PAM has a wide range of applications. The presence of amide groups on the side chains of PAM molecules gives them excellent water solubility and high reactivity, making them easy to obtain branched or network structures through grafting or crosslinking; The good viscoelasticity of PAM solution mainly comes from intermolecular interactions, molecular entanglement, and hydrogen bonding. Due to the significant impact of factors such as the degree of hydrolysis and modification methods of polymer molecules on their solution properties and application effects, studying the spatial configuration and intermolecular forces of polymer chain like macromolecules on solution properties and exploring the structure-activity relationship is of great significance.
The traditional method studies the related properties of polymer systems by exploring the rheological and viscoelastic properties of polymer solutions. Methods such as UV-Vis, SEM, light scattering, and chromatography have been applied to the study of polymer system configuration information. However, the above methods can only obtain the basic properties and configurations of polymer solutions, and cannot accurately describe the behavior and structure-activity relationship of molecules in polymer solutions at the micro scale. Computer simulation technology can qualitatively describe the process by establishing mathematical models and can simulate some quantitative relationships between the structure and performance of the system. Studying the molecular behavior of polymer systems at the micro scale is of great significance for analyzing polymer properties and predicting performance. This article reviews the research progress of the application of computer molecular simulation technology in PAM used in oil fields, and elaborates on the main molecular simulation technologies and application types.
1.Overview of Molecular Simulation
According to the different spatial and temporal scales, molecular simulation methods can mainly be divided into electronic scale, molecular (atomic) scale, and mesoscopic scale. The simulation at the electronic scale is mainly based on quantum mechanics methods. Quantum mechanics simulation is based on the study of the non localization of electrons in molecules, mainly including ab initio calculations, semi empirical molecular orbital calculations, and density functional theory. It can calculate most of the properties of molecules, such as conformation, ionization energy, electron density, transition states, and reaction pathways. The molecular (atomic) scale calculation methods mainly include molecular mechanics (MM), molecular dynamics (MD), and Monte Carlo calculation. They do not consider the motion and changes of electrons, and describe the interaction forces within and between molecules using atoms or molecular clusters as the smallest unit. Based on statistical theories such as ensemble averaging, problems can be analyzed. They can simulate the static structure and dynamic behavior of the system, obtain the equilibrium structure and thermodynamic properties of the molecule, but cannot obtain other properties related to electronic structure. The above simulation methods are all focused on microscale simulations, which may have limitations when dealing with multi-scale problems. The model of the mesoscale method is larger in both spatial and temporal scales than traditional methods, with the basic unit being beads composed of several functional groups. Therefore, the application of mesoscale simulation can simulate more complex systems, and can study complex fluids, polymer composite systems, and complex material systems at the nanoscale to micrometer scale. The calculation methods at the mesoscale include coarse-grained molecular dynamics (CGMD), dissipative particle dynamics (DPD), Brownian dynamics simulations, and lattice Boltzmann simulations.
Most studies on PAM systems aim to explore molecular structures or system properties, and relevant simulation methods based on classical mechanics are the best calculation methods. Discuss the structural changes of molecules through typical structural parameters and forces, and characterize bond length, bond angle, dihedral angle changes, and non bonding interactions in the form of mathematical functions. The main molecular simulation methods involved are: 1) MD. MD solves the time evolution state of the system based on Newton's laws of motion. Set the initial position, initial velocity, and molecular potential energy of each molecule. Through intermolecular forces, the initial position moves towards lower energy states, generating new molecular coordinates and momentum. The obtained result will be used as new data input, and the above steps will be repeated until equilibrium is reached. By integrating Newton's equations of motion, the dynamic information of the entire system can be obtained, including basic thermodynamic properties, mechanical properties, and radial distribution functions of liquids. MD has fast computing speed and high accuracy, but there are also limitations, such as harsh application conditions and limited time scale to the nanosecond level. 2) CGMD. CGMD ignores the intramolecular information in the system, treating several atoms or atomic clusters as a large coarse particle and defining them as coarse-grained particles, and then applying a force field for calculation. The basic principles and simulation techniques of CGMD are the same as traditional all atom/molecular dynamics, but currently there is no unified standard potential field, which is usually developed based on the specific situation of the studied system. 3) DPD. DPD is based on a collection of multiple atoms or molecules and is defined as "beads". The DPD model includes three pairs of interaction forces: conservative force, dissipative force, and random force. Through the damping effect of dissipative force on the motion of beads and the random collision effect generated by random force, the entire system maintains momentum balance during the reaction process, which has certain advantages in analyzing the interaction forces between particles in complex fluids.
2. Application of Molecular Simulation in PAM
2.1 Performance Analysis and Simulation of Polymer Systems
PAM has a good viscosity increasing effect, but viscosity loss is more severe in high salinity environments of oil reservoirs. There has been extensive research on the effect of inorganic salts on the viscosity of polymer solutions. In recent years, the use of molecular simulation technology to provide a detailed explanation of the effect of inorganic salts on the viscosity of polymer solutions from a microscopic perspective has attracted the attention of researchers. By studying the structural changes of polymer molecules under different mineralization levels through computer simulation, the macroscopic properties of polymers can be intuitively understood, which has important guiding significance for polymer molecule modification and synthesis.
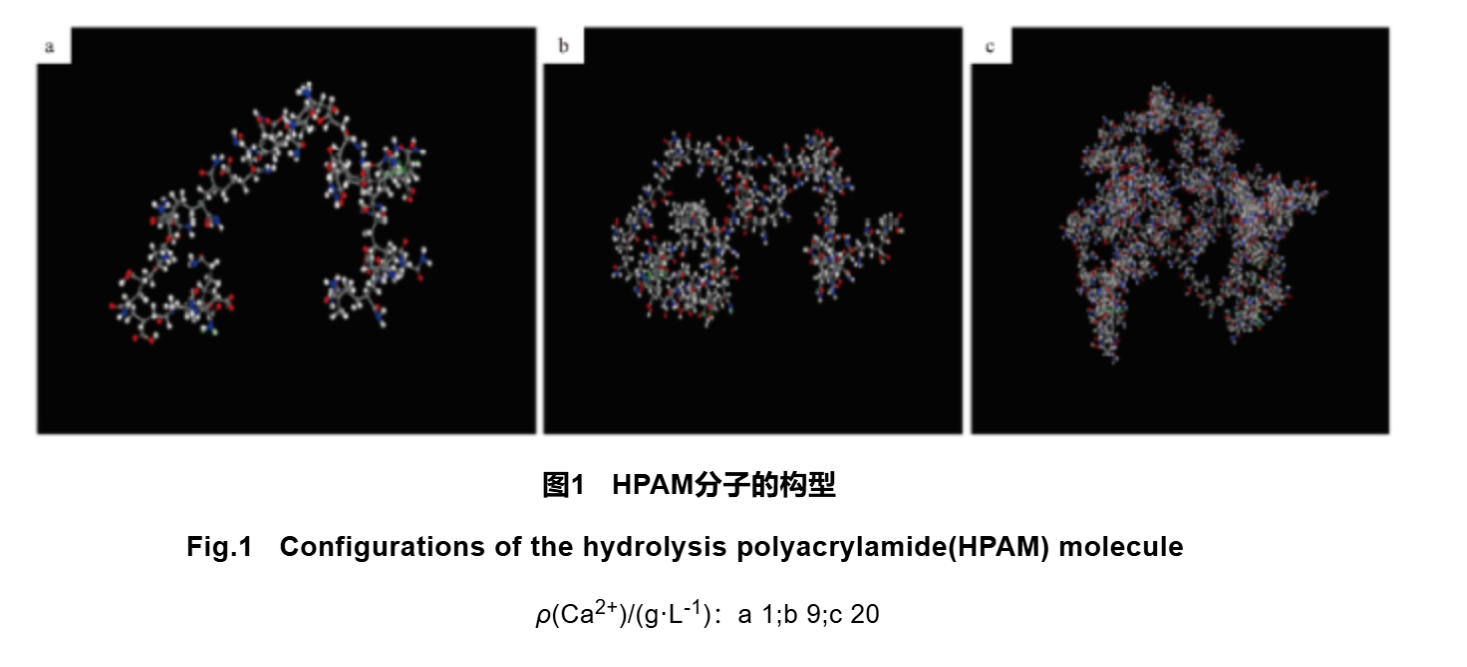
Jin Yanxin et al. used a combination of experimental and MD methods to study the effect of inorganic salts on the viscosity of hydrolyzed acrylamide (HPAM) solution. The particles in the simulated system were all subjected to a COMPASS force field. The experimental results indicate that the viscosity of polymer solutions is highly sensitive to inorganic salts, and the influence of MgCl2 and CaCl2 on solution viscosity is greater than that of NaCl. The simulation results indicate that there is an electrostatic attraction between Na+, Ca2+, Mg2+and negatively charged carboxylic acid groups. The ion layer formed by cations around the molecular chain can shield the repulsive effect between carboxylic acid groups in the molecular chain, causing the molecular chain to contract, thereby reducing the viscosity of the polymer solution. Polymer solutions are heterogeneous at the microscale, where polymer chains connected by cross-linking bonds expand and form a dense phase, while dispersed polymer molecules form a diluent phase. Yang et al. further studied the HPAM solution by dividing it into micro dense phase, medium phase, and dilution phase. They used MD method to investigate the molecular configuration of HPAM under high mineralization and the changes in viscosity of polymer solution from micro dense phase to dilution phase. The experimental results indicate that HPAM molecules in the micro dense phase are interconnected through electrostatic attraction to form a network structure, and the electrostatic attraction is three orders of magnitude higher than the dilution phase. Under high concentration of calcium ions, the electrostatic energy between HPAM molecules is significantly reduced, and salt ions form a network connection with HPAM molecules, disrupting the network structure between HPAM molecules in the dense phase. HPAM molecules tend to curl and gradually transition from a curved shape to a long spherical shape (Figure 1). The calculated apparent viscosity decreased by 37%, which is similar to the experimental results, further indicating that the network structure formed by electrostatic attraction in the dense phase is the key to solution viscosity.
Yao et al. introduced three different monomers into the polymer chain to obtain modified anionic PAM (HM-HPAM). Each of these monomers has a benzene ring on its side chain, and their structural differences lie in the length of the side chain and the number of oxygen atoms in the side chain. MD was used to study the kinetic parameters such as gyratory radius (Rg), hydrodynamic radius (RH), and intrinsic viscosity ([η]) of HPAM and three types of HM-HPAM in salt solutions with different ionic strengths. The simulation results show that compared with HPAM, the Rg, RH, [η] changes of HM-HPAM are not significant, and it has stronger salt resistance, and the salt resistance of HM-HPAM-1 to HM-HPAM-3 gradually increases. This is because the steric hindrance of monomers can reduce the curling degree of macromolecular chains, thereby improving the salt resistance of modified polymers. Therefore, introducing steric hindrance monomers into polymers for modified molecular design can improve the salt resistance of polymers.
Deng et al. investigated the rheological properties of hydrophobic associated acrylamide copolymer (HA-PAM) in salt solutions, systematically investigated the effects of polymer concentration, salt solution concentration, temperature, and shear rate on the rheological properties of the solution, and used DPD mesoscopic simulation method to describe the molecular behavior of the polymer in detail. Figure 2a shows the results of DPD simulation of polymer molecules in solution. From Figure 2a, it can be seen that hydrophobic groups aggregate to form hydrophobic water bodies; With the increase of shear rate, the hydrophobic interaction between side chains is enhanced, and the degree of polymer aggregation is enhanced. This indicates that an appropriate shear rate can promote the association of hydrophobic side chains. The influence of salt on the behavior of HA-PAM molecules was investigated at the microscopic level, as shown in Figure 2b. On the one hand, the shielding effect of electrolytes leads to macromolecular curling, reduced RH, weakened intermolecular entanglement, and a decrease in solution viscosity. On the other hand, an increase in salt concentration helps to enhance intermolecular hydrophobic binding. When the sodium chloride content is 0.4%~3.0% (w), HA-PAM will form very tight intramolecular hydrophobic microregions, and the viscosity of the solution will decrease again. Therefore, both reductions in viscosity of the solution are caused by the shielding effect of inorganic salts.
.png)